- ORGANIQUE (CHIMIE)
- ORGANIQUE (CHIMIE)En dehors des deux thèmes de recherche fondamentaux, la pierre philosophale et l’élixir, les alchimistes avaient réussi, grâce à des recettes plus ou moins rationnelles, à élaborer de nombreuses substances (qui sont, à quelques impuretés près, ce qu’on appelle aujourd’hui composés définis), dont quelques-unes avaient déjà des applications pratiques.Mais l’origine des matières premières mises en œuvre, tirées de l’air, de l’eau, de la terre ou de la matière vivante, n’avait pas retenu particulièrement l’attention; c’est Nicolas Lémery qui, dans son Cours de chymie (1690), distingue le premier la « chimie minérale», qui ne fait appel, au départ, qu’à la matière inerte, et la «chimie organique», qui puise ses sources dans le règne animal ou dans le règne végétal.Cette distinction était justifiée par le dogme de la «force vitale», seule cause mystérieuse censée être alors capable d’édifier in vivo des composés comme l’«alcool vini» (l’éthanol) ou l’acide acétique.Comme toutes les sciences de la matière, la chimie a vu successivement se développer ses aspects extractif, descriptif, explicatif. Les chimistes des XVIIe et XVIIIe siècles savaient du moins transformer l’une en l’autre deux substances quelconques extraites du règne vivant, par exemple oxyder in vitro l’alcool en acide acétique; mais ils avaient également isolé et décrit des substances que le monde vivant ne fournit point, tels le chlorure d’éthyle, résultant de l’action de l’acide muriatique (acide chlorhydrique) sur l’alcool, et l’éther dit sulfurique (oxyde d’éthyle), résultant de la déshydratation de l’alcool sous l’influence de l’huile de vitriol (acide sulfurique).Ces produits de transformation devenaient de plus en plus nombreux, et les substances directement extraites du règne vivant ne constituaient, vers 1850, qu’une faible minorité du domaine de la «chimie organique», dont l’unité n’était sauvegardée que par le respect du dogme de la force vitale.Berthelot, s’appuyant sur quelques «synthèses», à partir de matières extraites du monde inerte, de substances considérées jusqu’alors comme spécifiquement organiques: alcool, méthane, méthanol, benzène, etc., émit l’hypothèse que toute substance organique pouvait être synthétisée in vitro à partir d’éléments minéraux; l’avenir devait parfaitement justifier cette hypothèse.La barrière édifiée par Lémery tombait. Cependant, la «chimie organique» subsiste, mais il convient d’en modifier la définition. Il existe aujourd’hui une «chimie des substances naturelles», qui répond à la conception ancienne, et une chimie organique plus générale, qui englobe la quasi-totalité des substances renfermant du carbone, si l’on en excepte le carbone natif, les carbonates minéraux et les carbures métalliques.Les substances organiques naturelles renferment toujours du carbone, presque toujours de l’hydrogène, très souvent de l’oxygène, souvent de l’azote, parfois du soufre; en outre, les substances artificielles renferment, éventuellement, des halogènes et, avec des fréquences très variables, la totalité des autres éléments.1. Caractères fondamentauxLes liaisons organiques sont essentiellement covalentes ; seuls quelques dérivés organométalliques présentent un caractère salin, d’ailleurs très limité, l’anion restant covalent.En conséquence, les substances organiques sont, pour la plupart, des molécules ou des macromolécules ; cela implique des points de fusion et d’ébullition relativement bas ainsi que des solubilités élevées dans des solvants eux-mêmes organiques et, le plus fréquemment, une faible aquasolubilité, sauf pour les molécules riches en oxygène et en azote.La chimie organique est le domaine du métastable : un petit nombre de composés organiques résistent quelque temps à des températures voisines de 800 0C, la plupart sont détruits vers 400 0C, beaucoup dès 200 0C, certains ne sont stables qu’à très basse température. Deux seulement, le dioxyde de carbone (que la chimie minérale revendique parfois) et le méthane, sont thermodynamiquement stables, c’est-à-dire prennent naissance à partir des éléments lorsqu’on abaisse très lentement la température d’une valeur très élevée jusqu’à la température ambiante. Soumis à cette épreuve, les autres corps organiques se décomposent en ces deux composés stables, ou en eau, ammoniac, hydracides, carbone, azote, etc. Pratiquement, la pyrogénation des composés organiques conduit à des résultats plus compliqués, l’équilibre n’étant atteint qu’au bout d’un temps très long, et des produits de dégradation thermodynamiquement instables à la température ambiante, comme le monoxyde de carbone, subsistent parfois très longtemps.La chimie organique est dominée par la notion de fonction : en effet, le carbone, mieux que tout autre élément, est capable de contracter, avec lui-même, des covalences stables; il en résulte la possibilité d’édifier des chaînes purement carbonées, de longueur finie ou pratiquement indéfinie, linéaires, ramifiées ou cycliques.Quadrivalent, le carbone, sauf dans le cas particulier de macromolécules (diamant), doit satisfaire par d’autres éléments les valences non occupées dans le squelette carboné par des atomes de carbone. Si ces valences sont toutes occupées par des atomes d’hydrogène, on est en présence d’un hydrocarbure saturé acyclique ou cyclique; si l’une au moins est occupée par un « hétéroatome » (atome différent de C ou de H), on a affaire à une fonction.La variété considérable des hydrocarbures, le nombre des atomes d’hydrogène non équivalents que portent la plupart d’entre eux montrent qu’une même fonction peut apparaître sur les squelettes les plus divers aux places les plus diverses. Ces composés à même fonction sont dits homologues. On en connaît parfois, pour une même fonction, des dizaines de mille, et l’on conçoit que les propriétés physiques et chimiques évoluent de l’un à l’autre de façon quasi continue en conservant cependant un grand nombre de réactions communes qui constituent les propriétés de la fonction.Le carbone ainsi que ses partenaires plurivalents les plus communs sont les éléments les plus aptes à contracter des liaisons multiples. En ce qui concerne les hydrocarbures, au squelette d’un carbure saturé correspondent généralement plusieurs hydrocarbures non saturés; c’est ainsi qu’au méthylbutane:
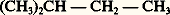 correspondent les carbures éthyléniques:
correspondent les carbures éthyléniques: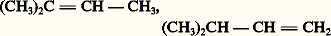 et le carbure acétylénique:
et le carbure acétylénique: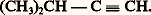 De plus, le carbure hypothétique de la formule 1 a ne peut exister sous cette forme; les doublets des doubles covalences ne sont pas localisés aux places indiquées, mais diffusent dans tout le squelette; il en résulte un édifice particulier, le benzène, représenté schématiquement en 1 b, qui est le prototype de toute une série d’hydrocarbures appelés benzénoïdes.Les hydrocarbures non saturés ou aromatiques peuvent porter des fonctions qui sont parfois différentes, quand elles affectent un carbone non saturé, des mêmes fonctions portées par un carbone saturé; c’est ainsi que la formule 2 a représente un alcool, mais la formule 2 b est celle d’un phénol.Des hétéroatomes peuvent participer à la formation de chaînes ouvertes ou fermées. Dans le premier cas, on a affaire à une fonction dérivée; c’est ainsi que:
De plus, le carbure hypothétique de la formule 1 a ne peut exister sous cette forme; les doublets des doubles covalences ne sont pas localisés aux places indiquées, mais diffusent dans tout le squelette; il en résulte un édifice particulier, le benzène, représenté schématiquement en 1 b, qui est le prototype de toute une série d’hydrocarbures appelés benzénoïdes.Les hydrocarbures non saturés ou aromatiques peuvent porter des fonctions qui sont parfois différentes, quand elles affectent un carbone non saturé, des mêmes fonctions portées par un carbone saturé; c’est ainsi que la formule 2 a représente un alcool, mais la formule 2 b est celle d’un phénol.Des hétéroatomes peuvent participer à la formation de chaînes ouvertes ou fermées. Dans le premier cas, on a affaire à une fonction dérivée; c’est ainsi que: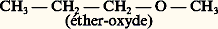 est une fonction dérivée de:
est une fonction dérivée de: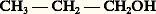 (et aussi de CH3OH). Dans le second cas, il s’agit d’un hétérocycle tel que l’éther-oxyde interne de la formule 3 (tétrahydro-furanne).Enfin, dans un «noyau benzénique», un ou deux groupes CH peuvent être remplacés par un hétéroatome de valence convenable: pyridine (formule 4 a), pyrrole (formule 4 b). Ce sont alors des «noyaux hétérocycliques».Sur un même hydrocarbure ou sur un même hétérocycle, on peut introduire des fonctions identiques ou différentes en très grand nombre. Seules des considérations pratiques, les difficultés rencontrées dans la synthèse ou l’isolement des molécules trop compliquées, s’opposent à la réalisation d’un édifice dont on s’impose la constitution.2. Les méthodes de la chimie organiqueLes principes de l’analyse immédiate sont développés dans d’autres articles (cf. CHROMATOGRAPHIE, DISTILLATION, ÉLECTROPHORÈSE).Identification d’une substance organiqueÀ l’origine surtout, le point de départ de l’identification d’une substance organique était l’analyse élémentaire. Elle permet de déterminer le pourcentage des divers éléments: C, H, O, N...; si la masse moléculaire, dont la cryoscopie fournit une valeur approchée, n’est pas trop grande, et si les résultats de l’analyse sont suffisamment précis, on en déduit la formule brute C 見H size=1廓O size=1塚N size=1嗀...Mais, sauf s’il s’agit de composés très simples, à cette formule correspondent un très grand nombre d’isomères entre lesquels il faut faire le départ; c’est le but de l’analyse fonctionnelle. Pendant longtemps, cette analyse a été purement chimique.À chaque fonction correspondent des propriétés dont certaines sont très caractéristiques: tous les alcools, notamment, sont transformés en alcoolates par le sodium. Mais la caractérisation de la fonction (ou des fonctions) ne suffit pas; il faut encore connaître la nature du squelette carboné et la place de la fonction sur ce squelette. Pendant très longtemps, le chimiste en était réduit à des méthodes de transformation menant à un composé antérieurement identifié; par exemple, la réduction de la plupart des composés organiques mène finalement à un hydrocarbure saturé ou cyclanique. Si celui-ci est connu, le squelette se trouve de ce fait établi. Quant à la place de la fonction sur ce squelette, elle peut être parfois déterminée par comparaison avec des homologues déjà identifiés; c’est ainsi qu’il existe des méthodes sûres pour distinguer les trois types de substitution d’un carbone hydroxylé (alcools primaires, secondaires, tertiaires).Mais ces méthodes sont défaillantes dans les cas compliqués; on a alors recours à des dégradations qui scindent la molécule primitive en divers tronçons plus faciles à identifier. Un cas très simple est celui d’un ester que l’hydrolyse coupe en un acide et en un alcool. Il est bon toutefois de vérifier les hypothèses structurales par une synthèse; sur l’exemple choisi, il faut montrer que l’acide et l’alcool engendrent bien, par perte d’eau, l’ester en question.Malheureusement, les exemples retenus sont beaucoup trop simples, et cette analyse sommaire ne reflète qu’imparfaitement les difficultés qu’ont rencontrées les chimistes.Le problème a été complètement transformé par l’introduction des contrôles physiques de structure: spectrographie ultraviolette, spectrographies infrarouge et Raman, résonance magnétique nucléaire (R.M.N.) et spectrographie de masse, pour ne citer que les plus utiles. Le spectre ultraviolet fournit de précieux renseignements sur l’existence et la nature des liaisons multiples, et le spectre infrarouge (ou Raman), plus général, présente des bandes caractéristiques de toutes les liaisons covalentes. Ces critères ne fournissent pas des renseignements absolus, car les physiciens ne peuvent calculer avec une précision suffisante les fréquences de ces bandes et surtout leurs variations avec la nature du squelette, mais la confrontation avec ces mêmes bandes observées sur des molécules plus simples, de même structure locale, fournit des renseignements inestimables. La résonance magnétique nucléaire, même réservée au proton, permet de compter les atomes d’hydrogène, de distinguer ceux qui jouent le même rôle et ceux qui jouent un rôle différent; toujours par référence à des composés connus de même structure locale, on peut identifier cette structure.La spectrographie de masse fournit une valeur très exacte de la masse moléculaire, mais aussi de la masse radicalaire des tronçons provenant de la rupture de la molécule. En admettant que deux molécules voisines ont les mêmes seuils de scission, on peut, par comparaison avec une molécule connue, reconstituer une partie de la structure.Ces renseignements ont permis d’établir complètement la structure de composés très compliqués, et ce sans analyse fonctionnelle chimique et même, parfois, sans analyse élémentaire. Cela vaut surtout pour la chimie des substances naturelles, car, s’il s’agit d’un produit synthétique, il est rare que l’on ne possède pas quelques présomptions sur sa structure.Parmi les «méthodes absolues», citons l’étude poussée des spectres de rayons X des molécules cristallisées, travail extrêmement laborieux et nécessitant l’emploi de calculettes, mais qui localise les divers atomes dans l’espace et ne laisse subsister aucun doute sur la structure.Le problème de l’identification des composés synthétiques est infiniment plus simple. Sauf anomalies imprévisibles, on ne peut guère hésiter qu’entre un très petit nombre de structures; s’il s’agit de choisir entre des formules non isomériques, une simple analyse quantitative permet de lever le litige. S’il s’agit d’isomères, la spectrographie ou la R.M.N. sont le plus souvent souveraines.Toutefois, nombreux sont les chimistes qui ne se contentent pas d’une détermination analytique, mais qui préfèrent une détermination synthétique. C’est ainsi que, depuis le début du XXe siècle, les chimistes ont mis un point d’honneur à reproduire synthétiquement des produits naturels dont la structure ne faisait plus de doute. S’il s’agit d’un produit synthétique, la confirmation de la structure consiste à réaliser la synthèse par une voie toute différente de celle qui lui avait donné naissance.SynthèsesEn principe, on appelle synthèse la préparation d’un corps composé à partir de corps simples, mais, surtout en chimie organique, l’extension abusive de ce terme en a fait presque le synonyme de «transformation»;On appellera «synthèses totales» celles qui répondent à la définition première: on peut citer, dans ce groupe, l’action de l’hydrogène sur le carbone, qui conduit, à très haute température, à l’acétylène C2H2 et, vers 1 000 0C, au méthane CH4.Très proche de ce type sont les synthèses «théoriquement totales», c’est-à-dire faisant appel, au départ, à des composés inorganiques dont la synthèse totale est possible; c’est ainsi que, lorsque le chimiste organicien emploie l’eau ou l’ammoniac à l’édification d’un composé, il n’est pas question pour lui de partir de l’hydrogène, de l’oxygène et de l’azote.Viennent ensuite les «synthèses partielles». Le point de départ peut être un composé organique emprunté au règne vivant, tel que l’alcool de fermentation. Cette synthèse est «théoriquement totale», puisque Berthelot a réussi la synthèse totale de l’éthanol. Dans d’autres cas, le chimiste fait appel à des produits naturels dont la synthèse totale n’a pas encore été réalisée; ces synthèses partielles deviendront théoriquement totales lorsque le produit de départ sera devenu «synthétique». Il est des domaines dans lesquels cet espoir semble vain; il est probable que la synthèse totale de la cellulose ne sera jamais possible; il n’en est pas moins vrai que la cellulose naturelle peut subir de nombreuses transformations, et l’acétylcellulose, par exemple, est un produit «semi-synthétique».Initialement, il s’agissait par la synthèse de détruire le dogme de la force vitale et de montrer que la biosynthèse repose sur des processus réactionnels sinon identiques, du moins comparables à ceux qui régissent les réactions effectuées in vitro. La différence essentielle réside dans la puissance et la haute spécificité des enzymes, mais celles-ci ne sont que des catalyseurs, certes compliqués, dont la synthèse totale paraît à l’heure actuelle irréalisable, bien qu’elle ait pu être réussie récemment dans quelques cas particuliers. Depuis Berthelot, la cause est entendue; un exemple de plus semble a priori n’apporter qu’une confirmation superflue, mais on conçoit que la reproduction artificielle d’un composé d’origine biologique tente encore le chimiste, ne serait-ce que pour en vérifier la structure encore incertaine, ou pour confirmer la puissance des techniques qui, de jour en jour, se perfectionnent. Rappelons seulement que la formule structurale du camphre n’a été définitivement acceptée que le jour où sa synthèse totale a pu être réussie.La synthèse a aussi pour but d’éprouver la généralité et les limites des réactions organiques, et surtout l’élaboration d’édifices complexes dont l’étude permet de confirmer ou d’infirmer les hypothèses encore fragiles sur lesquelles reposent nos conceptions sur la réactivité, en particulier d’étudier l’évolution des propriétés des fonctions avec la nature du squelette qui les porte. Elle a permis de montrer que quelques structures que la théorie élémentaire de la valence permettait de prévoir sont ou bien interdites, ou seulement stables aux très basses températures.L’aspect pratique de la synthèse est bien plus important encore. Beaucoup de produits, autrefois exclusivement naturels, présentent un gros intérêt, par eux-mêmes ou en tant que matières premières, pour la préparation de composés plus complexes. Il y a tout avantage à les préparer par synthèse totale ou partielle, que les sources naturelles soient insuffisantes ou que le prix de revient et la pureté d’un produit synthétique le rendent plus avantageux que le produit naturel. C’est ainsi que le méthanol, autrefois extrait péniblement des produits de pyrogénation du bois, est exclusivement préparé de nos jours à partir de l’oxyde de carbone et de l’hydrogène (synthèse totale), ou à partir du méthane naturel (synthèse partielle); la purification est alors beaucoup plus simple.La nature et le laboratoire entrent en concurrence pour d’autres produits: citons l’alcool éthylique qui provient en partie de la fermentation des jus sucrés, en partie d’une synthèse partielle à partir du pétrole, ou des gaz de hauts fourneaux.Mais la synthèse industrielle ne se borne pas à reproduire dans des conditions avantageuses des produits naturels. L’indigotier et la garance ont été longtemps les sources de deux excellents colorants, l’indigo et l’alizarine. Ceux-ci sont préparés synthétiquement à partir du goudron de houille, la culture de l’indigotier et de la garance n’étant plus rémunératrice. Mais, à côté des colorants naturels, la synthèse a mis à la disposition des teinturiers une gamme très étendue de matières colorantes nouvelles, adaptées aux diverses fibres naturelles et synthétiques. Les mêmes remarques s’appliquent dans le domaine des médicaments et des parfums.De plus, si de très rares produits naturels sont explosifs, aucun ne peut être employé comme tel, alors que de nombreux produits synthétiques constituent à la fois les explosifs brisants et les poudres propulsives.Pendant longtemps, des macromolécules naturelles (textiles, caoutchouc, colles, vernis) étaient seules employées en vue de ces usages respectifs, et, en dehors des bakélites (résines phénol-formol), toutes les résines synthétiques étaient considérées comme des sous-produits indésirables de réactions devant en principe conduire à des produits plus simples; depuis les choses ont complètement changé.Domestiquées, la haute polymérisation et la haute condensation conduisent à des macromolécules dépassant de loin, dans leur domaine, les produits naturels de même usage. Des textiles artificiels présentent toutes les qualités de la soie, avec une ténacité et une inaltérabilité bien supérieures; si le caoutchouc naturel est difficilement concurrencé par les caoutchoucs synthétiques en ce qui concerne l’élasticité, les élastomères synthétiques résistent mieux au vieillissement et à l’action des agents chimiques ; les colles synthétiques sont inodores et imputrescibles; les verres organiques sont plus légers, moins fragiles que les verres minéraux, et leurs éclats sont nettement moins tranchants.La synthèse des macromolécules est, une des branches les plus importantes de l’industrie organique.En dehors de quelques matières premières d’origine vivante, de moins en moins nombreuses, la synthèse industrielle fait appel à deux sources principales: la houille et le pétrole.La houille est le point de départ de la carbochimie ; la distillation de la houille fournit, d’une part, le goudron, source des carbures aromatiques et de leurs dérivés, d’autre part, le coke, matière première pour le monoxyde de carbone, l’hydrogène, l’acétylène.Le pétrole est la base de la pétrochimie. Le méthane naturel permet, en dehors de nombreuses synthèses, la préparation la plus économique de l’hydrogène; les termes simples du craquage: éthane, éthylène, propane, propylène isobutane et butènes, sont à la base des intermédiaires de synthèses les plus importants (oxyde d’éthylène, acétaldéhyde, acide acétique, glycol, glycérol...) et à base de macromolécules (polythène, élastomères, résines vinyliques...). Beaucoup de ces intermédiaires dérivaient autrefois de l’acétylène, donc de la carbochimie; le prix de revient élevé de l’acétylène, bien qu’il puisse également se préparer par pyrogénation du méthane, diminue de beaucoup son importance pratique.D’autre part, l’aromatisation des hydrocarbures moyens du pétrole donne un accès avantageux au toluène et à ses homologues. Si l’aromatisation de l’hexane en benzène n’est pas encore compétitive, on peut espérer qu’elle le sera un jour; dès lors, la carbochimie s’effacera à peu près complètement devant la pétrochimie.Néanmoins, des synthèses partielles restent intéressantes, tout au moins dans le domaine des parfums et des produits pharmaceutiques. C’est là que le mot « synthèse » a le plus perdu de sa signification originelle; par exemple, la transformation du cholestérol en hormones stéroïdes est couramment appelée «synthèse partielle»; or, ces hormones sont moins riches en atomes que le cholestérol de départ.Signalons enfin que la synthèse pratique a des limites. Des composés naturels aussi simples et aussi utiles que les sucres n’ont pu être reproduits artificiellement que récemment, et avec des rendements qui excluent tout espoir d’une synthèse concurrentielle. C’est peut-être moins certain en ce qui concerne les lipides; quant aux protides naturels, ils sont en général si complexes que leur synthèse pratique ne peut être envisagée.3. Les opérations de la chimie organiqueLa synthèse repose principalement sur deux opérations fondamentales: les transformations fonctionnelles et les réactions proprement synthétiques, c’est-à-dire qu’elles se traduisent par un allongement de la chaîne purement carbonée.Les modalités sont détaillées dans les articles relatifs aux diverses fonctions (ALCANES, ALCÈNES, etc.); on se bornera ici à quelques généralités.Les groupes fonctionnels peuvent être modifiés par oxydation ou par réduction; c’est ainsi que l’oxydation des alcools primaires ou secondaires mène à des dérivés carbonylés; réciproquement, ces derniers peuvent être hydrogénés en alcools.On peut également passer d’une fonction à une autre par substitution; c’est notamment le cas des alcools qui, sous l’influence des hydracides, sont transformés en éthers halohydriques.Deux autres opérations sont très générales: l’addition qui consiste en la fusion de deux radicaux sur deux atomes doublement ou triplement liés, par exemple l’addition d’un halogène à un carbure éthylénique ou celle d’un hydracide à un carbure acétylénique; l’opération inverse est l’élimination (déshydratation des alcools en alcènes).Les réactions proprement synthétiques présentent plus de variété. On peut citer des duplications, comme la réaction de Wurtz: 2 RI + 2 Na2 NaI + R 漣 R, et les additions des organométalliques sur les dérivés carbonylés; mais ce ne sont là que deux exemples d’un type beaucoup plus général.Il existe aussi des réactions de dégradations de la chaîne carbonée, qui décomposent une substance en molécules moins condensées en carbone; parfois préparatives, les dégradations ont surtout un intérêt dans la détermination des structures trop complexes pour pouvoir être établies directement.Sur des molécules de structure appropriée, ces opérations se traduisent éventuellement par des homocyclisations ou des hétérocyclisations ou par l’opération inverse, la décyclisation.Il existe enfin des réactions anormales, ou transpositions, qui diffèrent des opérations précédentes par un réarrangement des tronçons admis dans ces transformations: certaines constituent des préparations rémunératrices de composés définis.Il suffira ici de signaler la transposition qui accompagne généralement la déshydratation des glycols- 見 en dérivés carbonylés.Enfin, en chimie organique appliquée, les polymérisations et polycondensations, qui permettent de passer de un ou de plusieurs «monomères» à des macromolécules, ont pris une importance primordiale.4. Les théories de la chimie organiqueÀ peine promue au rang des véritables sciences après Lavoisier, la chimie organique devait connaître un essor prodigieux entre 1800 et 1900; de quelques dizaines de produits naturels elle passait à quelque 350 000 composés, dont la majorité provenaient de semi-synthèses. On comprendrait mal ces progrès si l’on faisait abstraction des théories qui les ont largement épaulés.Laissant de côté la théorie des types dont tout le mérite revient à Laurent et Gerhardt, on abordera tout d’abord la théorie de la valence. C’est un concept à la fois très général et très flou, commun à ses débuts à la chimie minérale et à la chimie organique: ainsi, l’univalence de l’hydrogène, du chlore, du sodium était admise tant par les minéralistes que par les organiciens. Cependant, la «valence des organiciens» repose sur des considérations toutes différentes de celles des minéralistes.Le point de départ est l’existence de l’isomérie. Si à la formule très simple CH4O correspond un seul composé défini, le méthanol, déjà à la formule C2H6O correspondent deux composés, l’éthanol et l’oxyde de méthyle, de propriétés physiques et chimiques totalement différentes; à une formule moyennement compliquée, telle que: C12H172N, correspondraient des centaines de composés très différents. On a donné le nom d’isomères à ces corps de même formule brute, mais nettement différents. Il était naturel de considérer que la molécule était alors constituée du même nombre des mêmes atomes, mais que l’enchaînement de ceux-ci était différent dans les divers isomères, ce qui entraîne la notion de «formule développée plane» (le mot plane spécifiant qu’il ne s’agit jusqu’ici que de l’ordre de succession des atomes et non de leur disposition spatiale et que la formule peut être décrite dans un plan). Il est démontré dans l’article STÉRÉOCHIMIE que la formule plane n’implique nullement l’unicité des composés qu’elle représente et qu’il est indispensable de recourir à la disposition spatiale des atomes; mais cela n’est nullement nécessaire pour introduire la notion de «valence des organiciens».Du point de vue historique, l’établissement des formules développées planes repose sur deux principes: l’existence des atomes, entités insécables, et la normalité des substitutions (absence de transpositions). Le cas de l’acide acétique est classique: c’est un composé ternaire C size=1見H size=1廓O size=1塚. L’oxyde d’argent lui enlève le quart de son hydrogène; 廓 est donc égal à 4 ou à un multiple de 4. Le chlore lui enlève successivement le quart, la moitié, les trois quarts de son hydrogène, alors que le quatrième quart disparaît encore sous l’action de l’oxyde d’argent, d’où la formule provisoire H3(C size=1見O size=1塚)H. Le perchlorure de phosphore lui enlève le quart de l’hydrogène et la moitié de l’oxygène; 塚 est donc égal à 2 ou à un multiple de 2, et cela fait pressentir le voisinage d’un hydrogène et d’un atome d’oxygène, d’où le schéma plus explicite H3(C size=1見O)OH. Enfin, la pyrogénation conduit au gaz carbonique et au méthane, d’où la présomption qu’il existe deux atomes de carbone et que deux atomes d’oxygène sont unis à l’un d’eux et au moins trois des atomes d’hydrogène à un autre atome de carbone, ce qui conduit à la formule provisoire 5 a. En représentant par des tirets les proximités des atomes, on arrive à la formule 5 b, très proche de celle universellement admise aujourd’hui.Il faut toutefois remarquer que l’on pouvait seulement écrire 廓 = 4n , 塚 = 2n , 見 = 2n , et non 廓 = 4, 塚 = 2, 見 = 2, et que la dernière réaction invoquée, la scission en méthane et en gaz carbonique, n’est pas, a priori, compatible avec la proximité d’un atome d’oxygène et d’un atome d’hydrogène.Or, le cas de l’acide acétique est celui qui a donné à ce genre d’analyse le minimum d’ambiguïté. On conçoit par là qu’il est impossible d’établir avec certitude une formule plane développée isolément, et que le système de ces formules ne se défend que par sa cohérence, c’est-à-dire qu’il rend compte le plus simplement possible des réactions qui font passer d’un composé à un autre; cela explique tous les tâtonnements indispensables avant d’arriver à ce système cohérent.Ce stade franchi, les chimistes du XIXe siècle furent attirés par une remarque d’importance fondamentale: dans toutes ces formules planes, l’hydrogène ne figurait jamais entre deux atomes, il était toujours en bout de chaîne; ils le considéraient donc comme univalent. L’oxygène pouvait, au maximum, unir deux atomes, l’azote trois, le carbone quatre. Les valences maximales de ces éléments étaient donc, respectivement, 2, 3 et 4 (en laissant de côté les sels d’ammonium qui, à l’époque, assignaient à l’azote la valence 5). Mais ces valences maximales n’étant pas toujours satisfaites, les chimistes du XIXe siècle remarquaient que la déficience de valence intéressait toujours deux atomes voisins; c’est ainsi qu’assignant à l’acétaldéhyde la formule CH3 漣 CH 漣 O ils constataient que le second carbone et l’oxygène y voyaient leur valence maximale diminuée d’une unité. Pour rétablir ces valences maximales, ils écrivaient entre C et O une liaison double, soit CH3 漣 CH 略O: telle est l’origine historique de la liaison double, qui comptait ainsi pour deux valences.Grâce à l’artifice des liaisons multiples, les valences des quatre éléments fondamentaux de la chimie organique, C, H, O, N, étaient respectivement 4, 1, 2, 3. Telles sont les «valences des organiciens».La chimie moderne a pu donner des interprétations plus précises de la nature des liaisons simples et multiples et corriger les quelques anomalies, explicables de nos jours par l’existence de liaisons semi-polaires; mais, telle quelle, la valence des organiciens a rendu d’immenses services.Elle a permis, en particulier, d’édifier les séquences d’atomes de carbone et de placer, sur ces hydrocarbures, les diverses fonctions, c’est-à-dire qu’elle a permis d’écrire toutes les formules planes compatibles avec une formule brute donnée et d’établir une systématique de la chimie organique.Très satisfaisante en ce qui concerne les structures, la valence n’était d’aucun secours pour l’interprétation des réactions.Les premières explications étaient des plus fantaisistes; on lit, dans les ouvrages anciens, que, si l’éthylate de sodium agit facilement sur le bromure d’éthyle:
(et aussi de CH3OH). Dans le second cas, il s’agit d’un hétérocycle tel que l’éther-oxyde interne de la formule 3 (tétrahydro-furanne).Enfin, dans un «noyau benzénique», un ou deux groupes CH peuvent être remplacés par un hétéroatome de valence convenable: pyridine (formule 4 a), pyrrole (formule 4 b). Ce sont alors des «noyaux hétérocycliques».Sur un même hydrocarbure ou sur un même hétérocycle, on peut introduire des fonctions identiques ou différentes en très grand nombre. Seules des considérations pratiques, les difficultés rencontrées dans la synthèse ou l’isolement des molécules trop compliquées, s’opposent à la réalisation d’un édifice dont on s’impose la constitution.2. Les méthodes de la chimie organiqueLes principes de l’analyse immédiate sont développés dans d’autres articles (cf. CHROMATOGRAPHIE, DISTILLATION, ÉLECTROPHORÈSE).Identification d’une substance organiqueÀ l’origine surtout, le point de départ de l’identification d’une substance organique était l’analyse élémentaire. Elle permet de déterminer le pourcentage des divers éléments: C, H, O, N...; si la masse moléculaire, dont la cryoscopie fournit une valeur approchée, n’est pas trop grande, et si les résultats de l’analyse sont suffisamment précis, on en déduit la formule brute C 見H size=1廓O size=1塚N size=1嗀...Mais, sauf s’il s’agit de composés très simples, à cette formule correspondent un très grand nombre d’isomères entre lesquels il faut faire le départ; c’est le but de l’analyse fonctionnelle. Pendant longtemps, cette analyse a été purement chimique.À chaque fonction correspondent des propriétés dont certaines sont très caractéristiques: tous les alcools, notamment, sont transformés en alcoolates par le sodium. Mais la caractérisation de la fonction (ou des fonctions) ne suffit pas; il faut encore connaître la nature du squelette carboné et la place de la fonction sur ce squelette. Pendant très longtemps, le chimiste en était réduit à des méthodes de transformation menant à un composé antérieurement identifié; par exemple, la réduction de la plupart des composés organiques mène finalement à un hydrocarbure saturé ou cyclanique. Si celui-ci est connu, le squelette se trouve de ce fait établi. Quant à la place de la fonction sur ce squelette, elle peut être parfois déterminée par comparaison avec des homologues déjà identifiés; c’est ainsi qu’il existe des méthodes sûres pour distinguer les trois types de substitution d’un carbone hydroxylé (alcools primaires, secondaires, tertiaires).Mais ces méthodes sont défaillantes dans les cas compliqués; on a alors recours à des dégradations qui scindent la molécule primitive en divers tronçons plus faciles à identifier. Un cas très simple est celui d’un ester que l’hydrolyse coupe en un acide et en un alcool. Il est bon toutefois de vérifier les hypothèses structurales par une synthèse; sur l’exemple choisi, il faut montrer que l’acide et l’alcool engendrent bien, par perte d’eau, l’ester en question.Malheureusement, les exemples retenus sont beaucoup trop simples, et cette analyse sommaire ne reflète qu’imparfaitement les difficultés qu’ont rencontrées les chimistes.Le problème a été complètement transformé par l’introduction des contrôles physiques de structure: spectrographie ultraviolette, spectrographies infrarouge et Raman, résonance magnétique nucléaire (R.M.N.) et spectrographie de masse, pour ne citer que les plus utiles. Le spectre ultraviolet fournit de précieux renseignements sur l’existence et la nature des liaisons multiples, et le spectre infrarouge (ou Raman), plus général, présente des bandes caractéristiques de toutes les liaisons covalentes. Ces critères ne fournissent pas des renseignements absolus, car les physiciens ne peuvent calculer avec une précision suffisante les fréquences de ces bandes et surtout leurs variations avec la nature du squelette, mais la confrontation avec ces mêmes bandes observées sur des molécules plus simples, de même structure locale, fournit des renseignements inestimables. La résonance magnétique nucléaire, même réservée au proton, permet de compter les atomes d’hydrogène, de distinguer ceux qui jouent le même rôle et ceux qui jouent un rôle différent; toujours par référence à des composés connus de même structure locale, on peut identifier cette structure.La spectrographie de masse fournit une valeur très exacte de la masse moléculaire, mais aussi de la masse radicalaire des tronçons provenant de la rupture de la molécule. En admettant que deux molécules voisines ont les mêmes seuils de scission, on peut, par comparaison avec une molécule connue, reconstituer une partie de la structure.Ces renseignements ont permis d’établir complètement la structure de composés très compliqués, et ce sans analyse fonctionnelle chimique et même, parfois, sans analyse élémentaire. Cela vaut surtout pour la chimie des substances naturelles, car, s’il s’agit d’un produit synthétique, il est rare que l’on ne possède pas quelques présomptions sur sa structure.Parmi les «méthodes absolues», citons l’étude poussée des spectres de rayons X des molécules cristallisées, travail extrêmement laborieux et nécessitant l’emploi de calculettes, mais qui localise les divers atomes dans l’espace et ne laisse subsister aucun doute sur la structure.Le problème de l’identification des composés synthétiques est infiniment plus simple. Sauf anomalies imprévisibles, on ne peut guère hésiter qu’entre un très petit nombre de structures; s’il s’agit de choisir entre des formules non isomériques, une simple analyse quantitative permet de lever le litige. S’il s’agit d’isomères, la spectrographie ou la R.M.N. sont le plus souvent souveraines.Toutefois, nombreux sont les chimistes qui ne se contentent pas d’une détermination analytique, mais qui préfèrent une détermination synthétique. C’est ainsi que, depuis le début du XXe siècle, les chimistes ont mis un point d’honneur à reproduire synthétiquement des produits naturels dont la structure ne faisait plus de doute. S’il s’agit d’un produit synthétique, la confirmation de la structure consiste à réaliser la synthèse par une voie toute différente de celle qui lui avait donné naissance.SynthèsesEn principe, on appelle synthèse la préparation d’un corps composé à partir de corps simples, mais, surtout en chimie organique, l’extension abusive de ce terme en a fait presque le synonyme de «transformation»;On appellera «synthèses totales» celles qui répondent à la définition première: on peut citer, dans ce groupe, l’action de l’hydrogène sur le carbone, qui conduit, à très haute température, à l’acétylène C2H2 et, vers 1 000 0C, au méthane CH4.Très proche de ce type sont les synthèses «théoriquement totales», c’est-à-dire faisant appel, au départ, à des composés inorganiques dont la synthèse totale est possible; c’est ainsi que, lorsque le chimiste organicien emploie l’eau ou l’ammoniac à l’édification d’un composé, il n’est pas question pour lui de partir de l’hydrogène, de l’oxygène et de l’azote.Viennent ensuite les «synthèses partielles». Le point de départ peut être un composé organique emprunté au règne vivant, tel que l’alcool de fermentation. Cette synthèse est «théoriquement totale», puisque Berthelot a réussi la synthèse totale de l’éthanol. Dans d’autres cas, le chimiste fait appel à des produits naturels dont la synthèse totale n’a pas encore été réalisée; ces synthèses partielles deviendront théoriquement totales lorsque le produit de départ sera devenu «synthétique». Il est des domaines dans lesquels cet espoir semble vain; il est probable que la synthèse totale de la cellulose ne sera jamais possible; il n’en est pas moins vrai que la cellulose naturelle peut subir de nombreuses transformations, et l’acétylcellulose, par exemple, est un produit «semi-synthétique».Initialement, il s’agissait par la synthèse de détruire le dogme de la force vitale et de montrer que la biosynthèse repose sur des processus réactionnels sinon identiques, du moins comparables à ceux qui régissent les réactions effectuées in vitro. La différence essentielle réside dans la puissance et la haute spécificité des enzymes, mais celles-ci ne sont que des catalyseurs, certes compliqués, dont la synthèse totale paraît à l’heure actuelle irréalisable, bien qu’elle ait pu être réussie récemment dans quelques cas particuliers. Depuis Berthelot, la cause est entendue; un exemple de plus semble a priori n’apporter qu’une confirmation superflue, mais on conçoit que la reproduction artificielle d’un composé d’origine biologique tente encore le chimiste, ne serait-ce que pour en vérifier la structure encore incertaine, ou pour confirmer la puissance des techniques qui, de jour en jour, se perfectionnent. Rappelons seulement que la formule structurale du camphre n’a été définitivement acceptée que le jour où sa synthèse totale a pu être réussie.La synthèse a aussi pour but d’éprouver la généralité et les limites des réactions organiques, et surtout l’élaboration d’édifices complexes dont l’étude permet de confirmer ou d’infirmer les hypothèses encore fragiles sur lesquelles reposent nos conceptions sur la réactivité, en particulier d’étudier l’évolution des propriétés des fonctions avec la nature du squelette qui les porte. Elle a permis de montrer que quelques structures que la théorie élémentaire de la valence permettait de prévoir sont ou bien interdites, ou seulement stables aux très basses températures.L’aspect pratique de la synthèse est bien plus important encore. Beaucoup de produits, autrefois exclusivement naturels, présentent un gros intérêt, par eux-mêmes ou en tant que matières premières, pour la préparation de composés plus complexes. Il y a tout avantage à les préparer par synthèse totale ou partielle, que les sources naturelles soient insuffisantes ou que le prix de revient et la pureté d’un produit synthétique le rendent plus avantageux que le produit naturel. C’est ainsi que le méthanol, autrefois extrait péniblement des produits de pyrogénation du bois, est exclusivement préparé de nos jours à partir de l’oxyde de carbone et de l’hydrogène (synthèse totale), ou à partir du méthane naturel (synthèse partielle); la purification est alors beaucoup plus simple.La nature et le laboratoire entrent en concurrence pour d’autres produits: citons l’alcool éthylique qui provient en partie de la fermentation des jus sucrés, en partie d’une synthèse partielle à partir du pétrole, ou des gaz de hauts fourneaux.Mais la synthèse industrielle ne se borne pas à reproduire dans des conditions avantageuses des produits naturels. L’indigotier et la garance ont été longtemps les sources de deux excellents colorants, l’indigo et l’alizarine. Ceux-ci sont préparés synthétiquement à partir du goudron de houille, la culture de l’indigotier et de la garance n’étant plus rémunératrice. Mais, à côté des colorants naturels, la synthèse a mis à la disposition des teinturiers une gamme très étendue de matières colorantes nouvelles, adaptées aux diverses fibres naturelles et synthétiques. Les mêmes remarques s’appliquent dans le domaine des médicaments et des parfums.De plus, si de très rares produits naturels sont explosifs, aucun ne peut être employé comme tel, alors que de nombreux produits synthétiques constituent à la fois les explosifs brisants et les poudres propulsives.Pendant longtemps, des macromolécules naturelles (textiles, caoutchouc, colles, vernis) étaient seules employées en vue de ces usages respectifs, et, en dehors des bakélites (résines phénol-formol), toutes les résines synthétiques étaient considérées comme des sous-produits indésirables de réactions devant en principe conduire à des produits plus simples; depuis les choses ont complètement changé.Domestiquées, la haute polymérisation et la haute condensation conduisent à des macromolécules dépassant de loin, dans leur domaine, les produits naturels de même usage. Des textiles artificiels présentent toutes les qualités de la soie, avec une ténacité et une inaltérabilité bien supérieures; si le caoutchouc naturel est difficilement concurrencé par les caoutchoucs synthétiques en ce qui concerne l’élasticité, les élastomères synthétiques résistent mieux au vieillissement et à l’action des agents chimiques ; les colles synthétiques sont inodores et imputrescibles; les verres organiques sont plus légers, moins fragiles que les verres minéraux, et leurs éclats sont nettement moins tranchants.La synthèse des macromolécules est, une des branches les plus importantes de l’industrie organique.En dehors de quelques matières premières d’origine vivante, de moins en moins nombreuses, la synthèse industrielle fait appel à deux sources principales: la houille et le pétrole.La houille est le point de départ de la carbochimie ; la distillation de la houille fournit, d’une part, le goudron, source des carbures aromatiques et de leurs dérivés, d’autre part, le coke, matière première pour le monoxyde de carbone, l’hydrogène, l’acétylène.Le pétrole est la base de la pétrochimie. Le méthane naturel permet, en dehors de nombreuses synthèses, la préparation la plus économique de l’hydrogène; les termes simples du craquage: éthane, éthylène, propane, propylène isobutane et butènes, sont à la base des intermédiaires de synthèses les plus importants (oxyde d’éthylène, acétaldéhyde, acide acétique, glycol, glycérol...) et à base de macromolécules (polythène, élastomères, résines vinyliques...). Beaucoup de ces intermédiaires dérivaient autrefois de l’acétylène, donc de la carbochimie; le prix de revient élevé de l’acétylène, bien qu’il puisse également se préparer par pyrogénation du méthane, diminue de beaucoup son importance pratique.D’autre part, l’aromatisation des hydrocarbures moyens du pétrole donne un accès avantageux au toluène et à ses homologues. Si l’aromatisation de l’hexane en benzène n’est pas encore compétitive, on peut espérer qu’elle le sera un jour; dès lors, la carbochimie s’effacera à peu près complètement devant la pétrochimie.Néanmoins, des synthèses partielles restent intéressantes, tout au moins dans le domaine des parfums et des produits pharmaceutiques. C’est là que le mot « synthèse » a le plus perdu de sa signification originelle; par exemple, la transformation du cholestérol en hormones stéroïdes est couramment appelée «synthèse partielle»; or, ces hormones sont moins riches en atomes que le cholestérol de départ.Signalons enfin que la synthèse pratique a des limites. Des composés naturels aussi simples et aussi utiles que les sucres n’ont pu être reproduits artificiellement que récemment, et avec des rendements qui excluent tout espoir d’une synthèse concurrentielle. C’est peut-être moins certain en ce qui concerne les lipides; quant aux protides naturels, ils sont en général si complexes que leur synthèse pratique ne peut être envisagée.3. Les opérations de la chimie organiqueLa synthèse repose principalement sur deux opérations fondamentales: les transformations fonctionnelles et les réactions proprement synthétiques, c’est-à-dire qu’elles se traduisent par un allongement de la chaîne purement carbonée.Les modalités sont détaillées dans les articles relatifs aux diverses fonctions (ALCANES, ALCÈNES, etc.); on se bornera ici à quelques généralités.Les groupes fonctionnels peuvent être modifiés par oxydation ou par réduction; c’est ainsi que l’oxydation des alcools primaires ou secondaires mène à des dérivés carbonylés; réciproquement, ces derniers peuvent être hydrogénés en alcools.On peut également passer d’une fonction à une autre par substitution; c’est notamment le cas des alcools qui, sous l’influence des hydracides, sont transformés en éthers halohydriques.Deux autres opérations sont très générales: l’addition qui consiste en la fusion de deux radicaux sur deux atomes doublement ou triplement liés, par exemple l’addition d’un halogène à un carbure éthylénique ou celle d’un hydracide à un carbure acétylénique; l’opération inverse est l’élimination (déshydratation des alcools en alcènes).Les réactions proprement synthétiques présentent plus de variété. On peut citer des duplications, comme la réaction de Wurtz: 2 RI + 2 Na2 NaI + R 漣 R, et les additions des organométalliques sur les dérivés carbonylés; mais ce ne sont là que deux exemples d’un type beaucoup plus général.Il existe aussi des réactions de dégradations de la chaîne carbonée, qui décomposent une substance en molécules moins condensées en carbone; parfois préparatives, les dégradations ont surtout un intérêt dans la détermination des structures trop complexes pour pouvoir être établies directement.Sur des molécules de structure appropriée, ces opérations se traduisent éventuellement par des homocyclisations ou des hétérocyclisations ou par l’opération inverse, la décyclisation.Il existe enfin des réactions anormales, ou transpositions, qui diffèrent des opérations précédentes par un réarrangement des tronçons admis dans ces transformations: certaines constituent des préparations rémunératrices de composés définis.Il suffira ici de signaler la transposition qui accompagne généralement la déshydratation des glycols- 見 en dérivés carbonylés.Enfin, en chimie organique appliquée, les polymérisations et polycondensations, qui permettent de passer de un ou de plusieurs «monomères» à des macromolécules, ont pris une importance primordiale.4. Les théories de la chimie organiqueÀ peine promue au rang des véritables sciences après Lavoisier, la chimie organique devait connaître un essor prodigieux entre 1800 et 1900; de quelques dizaines de produits naturels elle passait à quelque 350 000 composés, dont la majorité provenaient de semi-synthèses. On comprendrait mal ces progrès si l’on faisait abstraction des théories qui les ont largement épaulés.Laissant de côté la théorie des types dont tout le mérite revient à Laurent et Gerhardt, on abordera tout d’abord la théorie de la valence. C’est un concept à la fois très général et très flou, commun à ses débuts à la chimie minérale et à la chimie organique: ainsi, l’univalence de l’hydrogène, du chlore, du sodium était admise tant par les minéralistes que par les organiciens. Cependant, la «valence des organiciens» repose sur des considérations toutes différentes de celles des minéralistes.Le point de départ est l’existence de l’isomérie. Si à la formule très simple CH4O correspond un seul composé défini, le méthanol, déjà à la formule C2H6O correspondent deux composés, l’éthanol et l’oxyde de méthyle, de propriétés physiques et chimiques totalement différentes; à une formule moyennement compliquée, telle que: C12H172N, correspondraient des centaines de composés très différents. On a donné le nom d’isomères à ces corps de même formule brute, mais nettement différents. Il était naturel de considérer que la molécule était alors constituée du même nombre des mêmes atomes, mais que l’enchaînement de ceux-ci était différent dans les divers isomères, ce qui entraîne la notion de «formule développée plane» (le mot plane spécifiant qu’il ne s’agit jusqu’ici que de l’ordre de succession des atomes et non de leur disposition spatiale et que la formule peut être décrite dans un plan). Il est démontré dans l’article STÉRÉOCHIMIE que la formule plane n’implique nullement l’unicité des composés qu’elle représente et qu’il est indispensable de recourir à la disposition spatiale des atomes; mais cela n’est nullement nécessaire pour introduire la notion de «valence des organiciens».Du point de vue historique, l’établissement des formules développées planes repose sur deux principes: l’existence des atomes, entités insécables, et la normalité des substitutions (absence de transpositions). Le cas de l’acide acétique est classique: c’est un composé ternaire C size=1見H size=1廓O size=1塚. L’oxyde d’argent lui enlève le quart de son hydrogène; 廓 est donc égal à 4 ou à un multiple de 4. Le chlore lui enlève successivement le quart, la moitié, les trois quarts de son hydrogène, alors que le quatrième quart disparaît encore sous l’action de l’oxyde d’argent, d’où la formule provisoire H3(C size=1見O size=1塚)H. Le perchlorure de phosphore lui enlève le quart de l’hydrogène et la moitié de l’oxygène; 塚 est donc égal à 2 ou à un multiple de 2, et cela fait pressentir le voisinage d’un hydrogène et d’un atome d’oxygène, d’où le schéma plus explicite H3(C size=1見O)OH. Enfin, la pyrogénation conduit au gaz carbonique et au méthane, d’où la présomption qu’il existe deux atomes de carbone et que deux atomes d’oxygène sont unis à l’un d’eux et au moins trois des atomes d’hydrogène à un autre atome de carbone, ce qui conduit à la formule provisoire 5 a. En représentant par des tirets les proximités des atomes, on arrive à la formule 5 b, très proche de celle universellement admise aujourd’hui.Il faut toutefois remarquer que l’on pouvait seulement écrire 廓 = 4n , 塚 = 2n , 見 = 2n , et non 廓 = 4, 塚 = 2, 見 = 2, et que la dernière réaction invoquée, la scission en méthane et en gaz carbonique, n’est pas, a priori, compatible avec la proximité d’un atome d’oxygène et d’un atome d’hydrogène.Or, le cas de l’acide acétique est celui qui a donné à ce genre d’analyse le minimum d’ambiguïté. On conçoit par là qu’il est impossible d’établir avec certitude une formule plane développée isolément, et que le système de ces formules ne se défend que par sa cohérence, c’est-à-dire qu’il rend compte le plus simplement possible des réactions qui font passer d’un composé à un autre; cela explique tous les tâtonnements indispensables avant d’arriver à ce système cohérent.Ce stade franchi, les chimistes du XIXe siècle furent attirés par une remarque d’importance fondamentale: dans toutes ces formules planes, l’hydrogène ne figurait jamais entre deux atomes, il était toujours en bout de chaîne; ils le considéraient donc comme univalent. L’oxygène pouvait, au maximum, unir deux atomes, l’azote trois, le carbone quatre. Les valences maximales de ces éléments étaient donc, respectivement, 2, 3 et 4 (en laissant de côté les sels d’ammonium qui, à l’époque, assignaient à l’azote la valence 5). Mais ces valences maximales n’étant pas toujours satisfaites, les chimistes du XIXe siècle remarquaient que la déficience de valence intéressait toujours deux atomes voisins; c’est ainsi qu’assignant à l’acétaldéhyde la formule CH3 漣 CH 漣 O ils constataient que le second carbone et l’oxygène y voyaient leur valence maximale diminuée d’une unité. Pour rétablir ces valences maximales, ils écrivaient entre C et O une liaison double, soit CH3 漣 CH 略O: telle est l’origine historique de la liaison double, qui comptait ainsi pour deux valences.Grâce à l’artifice des liaisons multiples, les valences des quatre éléments fondamentaux de la chimie organique, C, H, O, N, étaient respectivement 4, 1, 2, 3. Telles sont les «valences des organiciens».La chimie moderne a pu donner des interprétations plus précises de la nature des liaisons simples et multiples et corriger les quelques anomalies, explicables de nos jours par l’existence de liaisons semi-polaires; mais, telle quelle, la valence des organiciens a rendu d’immenses services.Elle a permis, en particulier, d’édifier les séquences d’atomes de carbone et de placer, sur ces hydrocarbures, les diverses fonctions, c’est-à-dire qu’elle a permis d’écrire toutes les formules planes compatibles avec une formule brute donnée et d’établir une systématique de la chimie organique.Très satisfaisante en ce qui concerne les structures, la valence n’était d’aucun secours pour l’interprétation des réactions.Les premières explications étaient des plus fantaisistes; on lit, dans les ouvrages anciens, que, si l’éthylate de sodium agit facilement sur le bromure d’éthyle: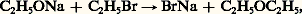 c’est parce que le sodium et le brome possèdent l’un pour l’autre une très grande affinité. On sait aujourd’hui que l’ion Na+ et l’ion Br- sont, pratiquement, sans affinité l’un pour l’autre, mais qu’au contraire les ions C2H5- et C2H+5 s’unissent énergiquement.Bien des «théories» anciennes ne sont que des règles empiriques, entre autres la «théorie des radicaux négatifs». Elle postule seulement que l’hydrogène lié à un carbone voisin de certains groupements est particulièrement apte à entrer en réaction.Empiriques également les «règles d’Hollemann» qui précisent les places de substitution sur les noyaux benzéniques.Il en est encore de même des «théories de l’affinité variable». Certains radicaux étaient censés accaparer une grande partie de l’affinité du carbone qui leur était directement relié, lui en laissant peu pour ses autres substituants, rendus aussi très mobiles: on attribuait à ces radicaux ambitieux une grande «capacité affinitaire»... En cas de compétition, certains radicaux quittaient plus volontiers que d’autres le carbone auquel ils étaient reliés: on leur attribuait une «aptitude migrative» élevée. En bonne logique, capacité affinitaire et aptitude migratrice devaient se classer en sens inverse; or ce n’est pas le cas, ce qui ruine cette hypothèse.En vérité, aucune théorie n’a présenté un aspect vraiment scientifique avant le développement de l’atomistique. Le point de départ fondamental est la nature de la rupture d’une liaison covalente. On admet aujourd’hui que cette liaison peut se couper de deux façons, soit en partageant le doublet covalent entre les deux radicaux résultants (rupture homolytique ou radicalaire), soit en laissant ce doublet à l’un des tronçons, qui devient un anion, alors que l’autre devient un cation (rupture hétérolytique ou ionique).Les réactions radicalaires sont fréquentes à haute température en phase gazeuse ou sous l’influence des radiations, de certains métaux et d’initiateurs de radicaux libres tels que les peroxydes. Les réactions hétérolytiques, plus nombreuses, sont la règle en phase liquide, et surtout en solution dans un milieu polaire. Les deux types de réactions ont fait l’objet de nombreux travaux théoriques qui ne peuvent être résumés ici (cf. RÉACTIONS CHIMIQUES).Qu’il suffise de signaler que la réactivité d’une fonction varie avec le squelette qui la porte selon des règles encore semi-empiriques qui font intervenir des transferts d’électrons. En tout cas, ces conceptions modernes ont permis d’interpréter plus scientifiquement les vieilles règles empiriques.Des efforts, très méritoires, sont faits de nos jours pour donner à ces règles un caractère plus quantitatif; ils s’appuient sur les acquisitions de la mécanique quantique ondulatoire et conduisent souvent à une interprétation satisfaisante des phénomènes observés; toutefois, les hypothèses à la base et la précision des calculs sont encore insuffisantes pour conduire à des prévisions solides. En d’autres termes, en dépit des immenses progrès de l’atomistique, la chimie organique semble être encore loin de devenir une science déductive, et l’heure n’est pas venue où la simple lecture d’une formule permettra de prévoir quantitativement les propriétés de la molécule qu’elle représente; fort heureusement, les prévisions qualitatives sont très souvent confirmées par l’expérience, mais elles nécessitent seulement une extrapolation à partir des composés connus de structure aussi voisine que possible de celle de la substance en question.5. Place de la chimie organique dans le monde industriel et scientifiqueMême en laissant de côté la métallurgie, la chimie, d’après la capitalisation boursière, représente plus de la moitié de l’activité industrielle mondiale; au sein de la chimie, la chimie organique est largement prioritaire, et la part des macromolécules organiques y est devenue prépondérante. En ce qui concerne les matières premières, les produits naturels interviennent pour moins de 10 p. 100 et leur importance diminue chaque année. La carbochimie, bien qu’elle soit en régression, intervient encore pour 30 p. 100 environ, tandis que la pétrochimie est déjà largement prioritaire.L’industrie organique travaille directement pour le consommateur, ou bien elle fournit des matières élaborées aux nombreuses industries qui en sont présentement tributaires.Pour commencer par l’aspect le moins humanitaire de ses débouchés, c’est en premier lieu à la chimie organique que l’art de la guerre, tout au moins conventionnelle, a recours: elle fournit à l’armée les poudres propulsives, les explosifs, les «gaz de combats».En revanche, elle procure à la pharmacopée la presque totalité des médicaments, la pharmacie chimique ayant peu à peu détrôné la pharmacie galénique.En ce qui concerne l’habillement, elle ajoute aux fibres naturelles de nombreuses fibres synthétiques dont certaines dérivent de transformations de la cellulose naturelle, et d’autres de synthèses à partir de la houille ou du pétrole; enfin, les colorants synthétiques ont complètement éclipsé tous les colorants naturels.La parfumerie utilise toujours les essences naturelles, mais aussi des produits synthétiques dont certains présentent une nuance inconnue parmi ces essences.L’ameublement utilise beaucoup les résines synthétiques imitant les cuirs et le bois, et bien plus résistantes aux agents atmosphériques.L’industrie automobile emploie des vernis synthétiques, et parfois des carrosseries en matière plastique; la chimie organique intervient encore, dans ce domaine, par les élastomères synthétiques: pneumatiques, tuyaux d’amenée des carburants, joints élastiques, revêtement des sièges.L’électrotechnique emprunte à la chimie organique ses isolants, et la photographie ses films.L’industrie alimentaire et l’emballage font appel aux films synthétiques.L’industrie chimique elle-même demande à la chimie organique les revêtements de réacteurs résistant aux acides minéraux.Les savons et les détergents sont des produits semi-synthétiques dont les usages sont innombrables, tant dans l’économie domestique que dans l’industrie.Les ignifuges et les imperméabilisants sont, pour la plupart, de nature organique; il en est de même de la totalité des solvants employés au dégraissage des tissus.La chimie organique est tributaire des autres sciences. Par l’intermédiaire de la thermodynamique et de la cinétique chimiques, depuis longtemps elle fait appel aux mathématiques, et plus directement depuis la théorie quantomécanique de la valence.Les recours à la physique sont plus importants. La physique donne une interprétation rationnelle des procédés d’analyse immédiate et permet de les perfectionner; mais c’est dans les contrôles physiques de structure que son apport est le plus précieux.Le règne vivant fournit encore à l’organicien quelques matières premières, mais celui-ci a fréquemment recours à l’action enzymatique pour réaliser in vitro des transformations de produits naturels ou synthétiques; réciproquement, la chimie organique fournit au physicien des matières premières indispensables à la réalisation de son équipement ou servant de base à ses études.Mais c’est évidemment à la biologie que la chimie organique rend les plus signalés services. La biochimie n’est qu’une chimie organique compliquée, et la vie n’est qu’une suite de transformations de matières organiques. De plus en plus, les techniques de la chimie organique permettent d’isoler et d’identifier les composés intervenant dans les phénomènes biologiques, d’en suivre la localisation chez l’être vivant, et les transformations éventuelles. Un biologiste moderne ne saurait progresser dans sa discipline sans un recours constant à la chimie organique.
c’est parce que le sodium et le brome possèdent l’un pour l’autre une très grande affinité. On sait aujourd’hui que l’ion Na+ et l’ion Br- sont, pratiquement, sans affinité l’un pour l’autre, mais qu’au contraire les ions C2H5- et C2H+5 s’unissent énergiquement.Bien des «théories» anciennes ne sont que des règles empiriques, entre autres la «théorie des radicaux négatifs». Elle postule seulement que l’hydrogène lié à un carbone voisin de certains groupements est particulièrement apte à entrer en réaction.Empiriques également les «règles d’Hollemann» qui précisent les places de substitution sur les noyaux benzéniques.Il en est encore de même des «théories de l’affinité variable». Certains radicaux étaient censés accaparer une grande partie de l’affinité du carbone qui leur était directement relié, lui en laissant peu pour ses autres substituants, rendus aussi très mobiles: on attribuait à ces radicaux ambitieux une grande «capacité affinitaire»... En cas de compétition, certains radicaux quittaient plus volontiers que d’autres le carbone auquel ils étaient reliés: on leur attribuait une «aptitude migrative» élevée. En bonne logique, capacité affinitaire et aptitude migratrice devaient se classer en sens inverse; or ce n’est pas le cas, ce qui ruine cette hypothèse.En vérité, aucune théorie n’a présenté un aspect vraiment scientifique avant le développement de l’atomistique. Le point de départ fondamental est la nature de la rupture d’une liaison covalente. On admet aujourd’hui que cette liaison peut se couper de deux façons, soit en partageant le doublet covalent entre les deux radicaux résultants (rupture homolytique ou radicalaire), soit en laissant ce doublet à l’un des tronçons, qui devient un anion, alors que l’autre devient un cation (rupture hétérolytique ou ionique).Les réactions radicalaires sont fréquentes à haute température en phase gazeuse ou sous l’influence des radiations, de certains métaux et d’initiateurs de radicaux libres tels que les peroxydes. Les réactions hétérolytiques, plus nombreuses, sont la règle en phase liquide, et surtout en solution dans un milieu polaire. Les deux types de réactions ont fait l’objet de nombreux travaux théoriques qui ne peuvent être résumés ici (cf. RÉACTIONS CHIMIQUES).Qu’il suffise de signaler que la réactivité d’une fonction varie avec le squelette qui la porte selon des règles encore semi-empiriques qui font intervenir des transferts d’électrons. En tout cas, ces conceptions modernes ont permis d’interpréter plus scientifiquement les vieilles règles empiriques.Des efforts, très méritoires, sont faits de nos jours pour donner à ces règles un caractère plus quantitatif; ils s’appuient sur les acquisitions de la mécanique quantique ondulatoire et conduisent souvent à une interprétation satisfaisante des phénomènes observés; toutefois, les hypothèses à la base et la précision des calculs sont encore insuffisantes pour conduire à des prévisions solides. En d’autres termes, en dépit des immenses progrès de l’atomistique, la chimie organique semble être encore loin de devenir une science déductive, et l’heure n’est pas venue où la simple lecture d’une formule permettra de prévoir quantitativement les propriétés de la molécule qu’elle représente; fort heureusement, les prévisions qualitatives sont très souvent confirmées par l’expérience, mais elles nécessitent seulement une extrapolation à partir des composés connus de structure aussi voisine que possible de celle de la substance en question.5. Place de la chimie organique dans le monde industriel et scientifiqueMême en laissant de côté la métallurgie, la chimie, d’après la capitalisation boursière, représente plus de la moitié de l’activité industrielle mondiale; au sein de la chimie, la chimie organique est largement prioritaire, et la part des macromolécules organiques y est devenue prépondérante. En ce qui concerne les matières premières, les produits naturels interviennent pour moins de 10 p. 100 et leur importance diminue chaque année. La carbochimie, bien qu’elle soit en régression, intervient encore pour 30 p. 100 environ, tandis que la pétrochimie est déjà largement prioritaire.L’industrie organique travaille directement pour le consommateur, ou bien elle fournit des matières élaborées aux nombreuses industries qui en sont présentement tributaires.Pour commencer par l’aspect le moins humanitaire de ses débouchés, c’est en premier lieu à la chimie organique que l’art de la guerre, tout au moins conventionnelle, a recours: elle fournit à l’armée les poudres propulsives, les explosifs, les «gaz de combats».En revanche, elle procure à la pharmacopée la presque totalité des médicaments, la pharmacie chimique ayant peu à peu détrôné la pharmacie galénique.En ce qui concerne l’habillement, elle ajoute aux fibres naturelles de nombreuses fibres synthétiques dont certaines dérivent de transformations de la cellulose naturelle, et d’autres de synthèses à partir de la houille ou du pétrole; enfin, les colorants synthétiques ont complètement éclipsé tous les colorants naturels.La parfumerie utilise toujours les essences naturelles, mais aussi des produits synthétiques dont certains présentent une nuance inconnue parmi ces essences.L’ameublement utilise beaucoup les résines synthétiques imitant les cuirs et le bois, et bien plus résistantes aux agents atmosphériques.L’industrie automobile emploie des vernis synthétiques, et parfois des carrosseries en matière plastique; la chimie organique intervient encore, dans ce domaine, par les élastomères synthétiques: pneumatiques, tuyaux d’amenée des carburants, joints élastiques, revêtement des sièges.L’électrotechnique emprunte à la chimie organique ses isolants, et la photographie ses films.L’industrie alimentaire et l’emballage font appel aux films synthétiques.L’industrie chimique elle-même demande à la chimie organique les revêtements de réacteurs résistant aux acides minéraux.Les savons et les détergents sont des produits semi-synthétiques dont les usages sont innombrables, tant dans l’économie domestique que dans l’industrie.Les ignifuges et les imperméabilisants sont, pour la plupart, de nature organique; il en est de même de la totalité des solvants employés au dégraissage des tissus.La chimie organique est tributaire des autres sciences. Par l’intermédiaire de la thermodynamique et de la cinétique chimiques, depuis longtemps elle fait appel aux mathématiques, et plus directement depuis la théorie quantomécanique de la valence.Les recours à la physique sont plus importants. La physique donne une interprétation rationnelle des procédés d’analyse immédiate et permet de les perfectionner; mais c’est dans les contrôles physiques de structure que son apport est le plus précieux.Le règne vivant fournit encore à l’organicien quelques matières premières, mais celui-ci a fréquemment recours à l’action enzymatique pour réaliser in vitro des transformations de produits naturels ou synthétiques; réciproquement, la chimie organique fournit au physicien des matières premières indispensables à la réalisation de son équipement ou servant de base à ses études.Mais c’est évidemment à la biologie que la chimie organique rend les plus signalés services. La biochimie n’est qu’une chimie organique compliquée, et la vie n’est qu’une suite de transformations de matières organiques. De plus en plus, les techniques de la chimie organique permettent d’isoler et d’identifier les composés intervenant dans les phénomènes biologiques, d’en suivre la localisation chez l’être vivant, et les transformations éventuelles. Un biologiste moderne ne saurait progresser dans sa discipline sans un recours constant à la chimie organique.
Encyclopédie Universelle. 2012.